腺嘌呤去胺酶缺乏症
腺苷脫氨酶缺乏症(英語:Adenosine deaminase deficiency)缺乏症是一種體染色體隱性遺傳疾病 ,會破壞免疫系統並導致嚴重複合型免疫缺乏症(SCID)。 此病的病因是腺苷脫氨酶(ADA)缺乏導致核酸代謝產物的異常累積,令T、B淋巴細胞發育不全,功能障礙,引致嚴重的細胞、體液免疫缺陷。
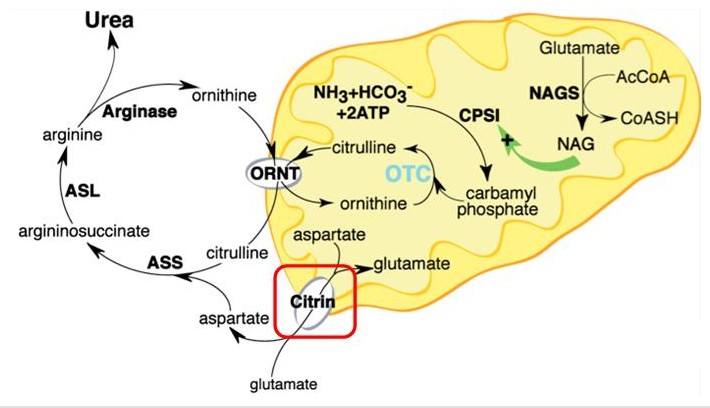
病因學存在人體中的氮主要由蛋白質和氨基酸代謝產生,但血氨是一種對人體有毒害的物質。因此肝臟會進行尿素代謝反應,把血氨轉換成較無毒性的尿素,排放到尿液中。而Citrin(見下圖紅框)為參與尿素代謝循環中的蛋白質之一,主要負責將Aspartate從粒線體運送至細胞質,讓Aspartate與Citrulline(瓜胺酸)藉由酵素Argininosuccinate synthase (ASS)形成Argininosuccinate(反應見下圖)。缺乏Citrin這個蛋白質,上述的反應及尿素代謝循環便 遭到阻斷,因而引發瓜胺酸血症及高血氨症等臨床症狀。Citrin缺乏症可分成兩種的臨床症狀表現:第二型瓜胺酸血症(citrullinemia type II,簡稱CTLN2)及新生兒膽汁鬱積症(neonatal intrahepatic cholestasis caused by citrin deficiency, 簡稱NICCD)。
發生率由於此疾病最早在日本被發現,先前一直認為Citrin缺乏症只發生在日本地區,但後來在其他國家也陸續有病例被報導。2005年,日本針對日本本國及其他鄰近亞洲國家的Citrin突變基因研究論文中統計發表了此突變基因在日本及中國(中國黃河以南包含台灣)及韓國的帶因率:日本:1/65、中國:1/65、中國南部(包含台灣):1/48、韓國:1/112。目前根據統計,台灣地區的發生率約一萬到兩萬分之一之間。遺傳模式Citrin缺乏症屬於體染色體隱性遺傳。各帶一個突變基因的父母(帶因者)會有25%的機率生下患病的子代,50%的機率生下無症狀的帶因者,25%的機率生下不帶有此突變基因的子代。臨床表徵Citrin缺乏症可分成兩種的臨床症狀表現:第二型瓜胺酸血症(citrullinemia type II, 簡稱CTLN2)及新生兒膽汁鬱積症(neonatal intrahepatic cholestasis caused by citrin deficiency, 簡稱NICCD)。部分的新生兒膽汁鬱積症NICCD患者會在成年時期發展成第二型瓜胺酸血症CTLN2。新生兒膽汁鬱積症(NICCD)由進展到第二型瓜胺酸血症(CTLN2)的過程是漸進且緩慢的,病程發展可見下圖。
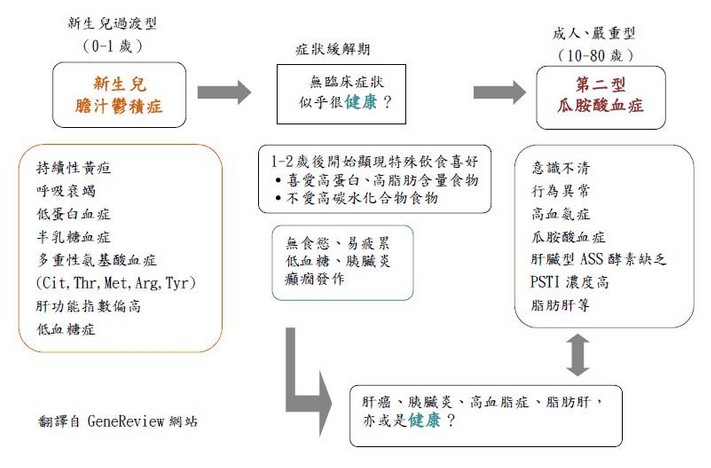
新生兒膽汁鬱積症(NICCD)通常發生在一歲之前,患者會有過度性的膽汁鬱積、脂肪肝、肝細胞纖維化、出生體重較輕、生長遲緩、低蛋白血症、凝血因子降低、溶血性貧血、肝腫大、程度不一的肝功能異常,也可能伴隨低血糖症。但患者的症狀 通常不太嚴重,而且在適當的治療下這些症狀通常在一歲後便消失。約二歲後,這些兒童開始顯現出對於高蛋白及高脂肪食物的特殊愛好,並且對於高糖份及高碳水化合物的食物的厭惡。約十年或二十年過後,部分個案會發展成嚴重型的第二型瓜胺酸血症(CTLN2)並出現神經精神症狀。
第二型瓜胺酸血症(CTLN2)主要在成年時期發病,發病年齡通常介於20-50歲之間。症狀表現主要是因為反覆性的高血氨引發的神經精神症狀如:易怒、過動、妄想、嗜睡、失憶、抽痙性痙攣、昏迷等,而患者可能因為腦部水腫而死亡。患者的肝臟切片會發現脂肪肝及輕度的纖維化,但其肝功能 並無異常,或只有輕度異常
病因學存在人體中的氮主要由蛋白質和氨基酸代謝產生,但血氨是一種對人體有毒害的物質。因此肝臟會進行尿素代謝反應,把血氨轉換成較無毒性的尿素,排放到尿液中。而Citrin(見下圖紅框)為參與尿素代謝循環中的蛋白質之一,主要負責將Aspartate從粒線體運送至細胞質,讓Aspartate與Citrulline(瓜胺酸)藉由酵素Argininosuccinate synthase (ASS)形成Argininosuccinate(反應見下圖)。缺乏Citrin這個蛋白質,上述的反應及尿素代謝循環便 遭到阻斷,因而引發瓜胺酸血症及高血氨症等臨床症狀。Citrin缺乏症可分成兩種的臨床症狀表現:第二型瓜胺酸血症(citrullinemia type II,簡稱CTLN2)及新生兒膽汁鬱積症(neonatal intrahepatic cholestasis caused by citrin deficiency, 簡稱NICCD)。
發生率由於此疾病最早在日本被發現,先前一直認為Citrin缺乏症只發生在日本地區,但後來在其他國家也陸續有病例被報導。2005年,日本針對日本本國及其他鄰近亞洲國家的Citrin突變基因研究論文中統計發表了此突變基因在日本及中國(中國黃河以南包含台灣)及韓國的帶因率:日本:1/65、中國:1/65、中國南部(包含台灣):1/48、韓國:1/112。目前根據統計,台灣地區的發生率約一萬到兩萬分之一之間。遺傳模式Citrin缺乏症屬於體染色體隱性遺傳。各帶一個突變基因的父母(帶因者)會有25%的機率生下患病的子代,50%的機率生下無症狀的帶因者,25%的機率生下不帶有此突變基因的子代。臨床表徵Citrin缺乏症可分成兩種的臨床症狀表現:第二型瓜胺酸血症(citrullinemia type II, 簡稱CTLN2)及新生兒膽汁鬱積症(neonatal intrahepatic cholestasis caused by citrin deficiency, 簡稱NICCD)。部分的新生兒膽汁鬱積症NICCD患者會在成年時期發展成第二型瓜胺酸血症CTLN2。新生兒膽汁鬱積症(NICCD)由進展到第二型瓜胺酸血症(CTLN2)的過程是漸進且緩慢的,病程發展可見下圖。
新生兒膽汁鬱積症(NICCD)通常發生在一歲之前,患者會有過度性的膽汁鬱積、脂肪肝、肝細胞纖維化、出生體重較輕、生長遲緩、低蛋白血症、凝血因子降低、溶血性貧血、肝腫大、程度不一的肝功能異常,也可能伴隨低血糖症。但患者的症狀 通常不太嚴重,而且在適當的治療下這些症狀通常在一歲後便消失。約二歲後,這些兒童開始顯現出對於高蛋白及高脂肪食物的特殊愛好,並且對於高糖份及高碳水化合物的食物的厭惡。約十年或二十年過後,部分個案會發展成嚴重型的第二型瓜胺酸血症(CTLN2)並出現神經精神症狀。
第二型瓜胺酸血症(CTLN2)主要在成年時期發病,發病年齡通常介於20-50歲之間。症狀表現主要是因為反覆性的高血氨引發的神經精神症狀如:易怒、過動、妄想、嗜睡、失憶、抽痙性痙攣、昏迷等,而患者可能因為腦部水腫而死亡。患者的肝臟切片會發現脂肪肝及輕度的纖維化,但其肝功能 並無異常,或只有輕度異常